We’d like to first thank Jon for the opportunity to discuss our work in this forum. We recently published a study investigating direct functional annotation of short metagenomic reads that stemmed from protocol development for our lab. Jon invited us to write a blog post on the subject, and we thought it would be a great venue to discuss some practical applications of our work and to share with the research community the motivation for our study and how it came about.
Our lab, the Borenstein Lab at the University of Washington, is broadly interested in metabolic modeling of the human microbiome (see, for example our Metagenomic Systems Biology approach) and in the development of novel computational methods for analyzing functional metagenomic data (see, for example, Metagenomic Deconvolution). In this capacity, we often perform large-scale analysis of publicly available metagenomic datasets as well as collaborate with experimental labs to analyze new metagenomic datasets, and accordingly we have developed extensive expertise in performing functional, community-level annotation of metagenomic samples. We focused primarily on protocols that derive functional profiles directly from short sequencing reads (e.g., by mapping the short reads to a collection of annotated genes), as such protocols provide gene abundance profiles that are relatively unbiased by species abundance in the sample or by the availability of closely-related reference genomes. Such functional annotation protocols are extremely common in the literature and are essential when approaching metagenomics from a gene-centric point of view, where the goal is to describe the community as a whole.
However, when we began to design our in-house annotation pipeline, we pored over the literature and realized that each research group and each metagenomic study applied a slightly different approach to functional annotation. When we implemented and evaluated these methods in the lab, we also discovered that the functional profiles obtained by the various methods often differ significantly. Discussing these findings with colleagues, some further expressed doubt that that such short sequencing reads even contained enough information to map back unambiguously to the correct function. Perhaps the whole approach was wrong!
We therefore set out to develop a set of ‘best practices’ for our lab for metagenomic sequence annotation and to prove (or disprove) quantitatively that such direct functional annotation of short reads provides a valid functional representation of the sample. We specifically decided to pursue a large-scale study, performed as rigorously as possible, taking into account both the phylogeny of the microbes in the sample and the phylogenetic coverage of the database, as well as several technical aspects of sequencing like base-calling error and read length. We have found this evaluation approach and the results we obtained quite useful for designing our lab protocols, and thought it would be helpful to share them with the wider metagenomics and microbiome research community. The result is our recent paper in PLoS One, Comparative Analysis of Functional Metagenomic Annotation and the Mappability of Short Reads.
To perform a rigorous study of functional annotation, we needed a set of reads whose true annotations were known (a “ground truth”). In other words, we had to know the exact locus and the exact genome from which each sequencing read originated and the functional classification associated with this locus. We further wanted to have complete control over technical sources of error. To accomplish this, we chose to implement a simulation scheme, deriving a huge collection of sequence reads from fully sequenced, well annotated, and curated genomes. This schemed allowed us to have complete information about the origin of each read and allowed us to simulate various technical factors we were interested in. Moreover, simulating sequencing reads allowed us to systematically eliminate variations in annotation performance due to technological or biological effects that would typically be convoluted in an experimental setup. For a set of curated genomes, we settled on the KEGG database, as it contained a large collection of consistently functionally curated microbial genomes and it has been widely used in metagenomics for sample annotation. The KEGG hierarchy of KEGG Orthology groups (KOs), Modules, and Pathways could then serve as a common basis for comparative analysis. To control for phylogenetic bias in our results, we sampled broadly across 23 phyla and 89 genera in the bacterial and archaeal tree of life, using a randomly selected strain in KEGG for each tip of the tree from Ciccarelli et al. From each of the selected 170 strains, we generated either *every* possible contiguous sequence of a given length or (in some cases) every 10th contiguous sequence, using a sliding window approach. We additionally introduced various models to simulate sequencing errors. This large collection of reads (totaling ~16Gb) were then aligned to the KEGG genes database using a translated BLAST mapping. To control for phylogenetic coverage of the database (the phylogenetic relationship of the database to the sequence being aligned) we also simulated mapping to many partial collections of genomes. We further used four common protocols from the literature to convert the obtained BLAST alignments to functional annotations. Comparing the resulting annotation of each read to the annotation of the gene from which it originated allowed us to systematically evaluate the accuracy of this annotation approach and to examine the effect of various factors, including read length, sequencing error, and phylogeny.
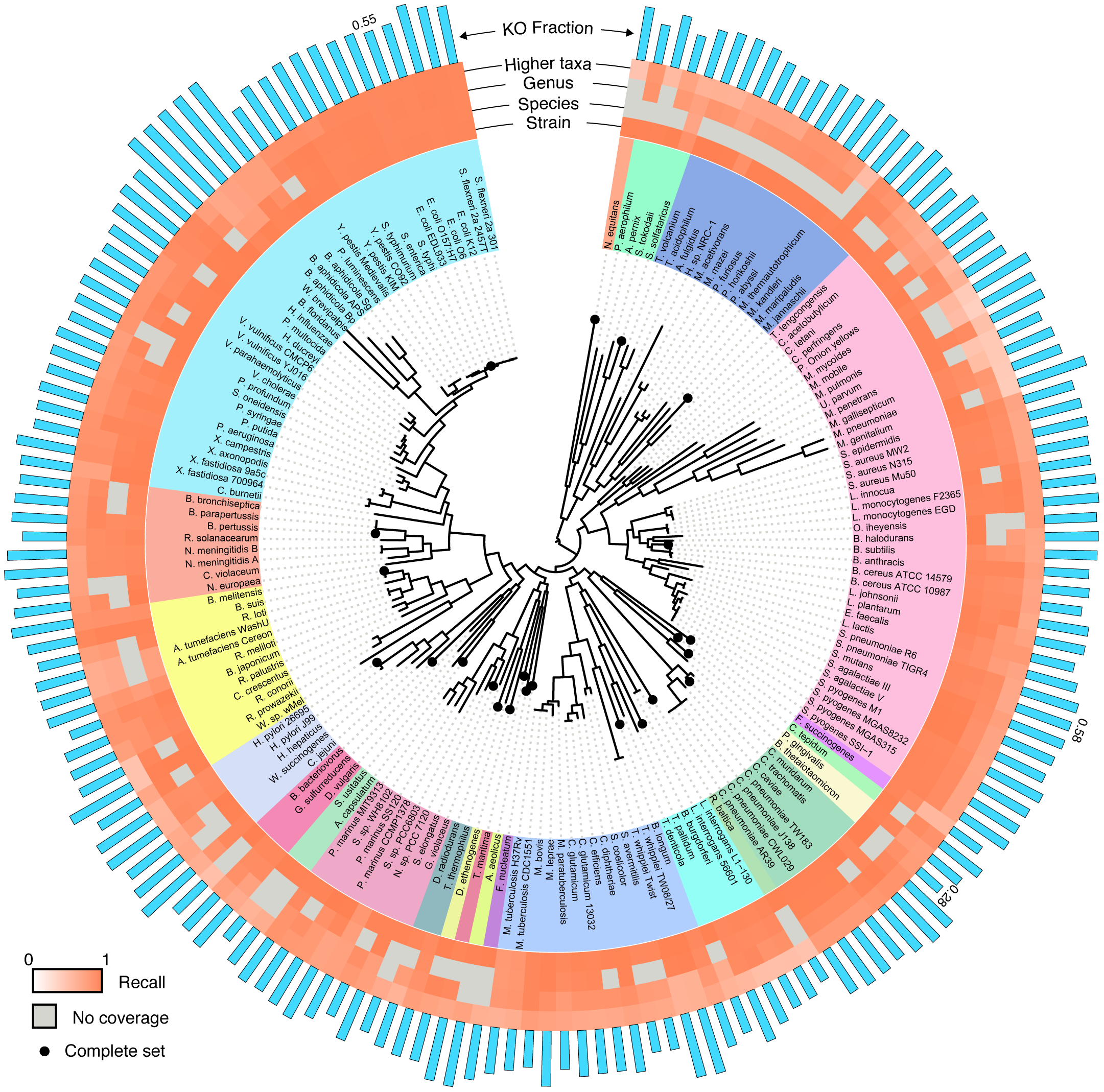
First and foremost, we confirmed that direct annotation of short reads indeed provides an overall accurate functional description of both individual reads and the sample as a whole. In other words, short reads appear to contain enough information to identify the functional annotation of the gene they originated from (although, not necessarily the specific taxa of origin). Functions of individual reads were identified with high precision and recall, yet the recall was found to be clade dependent. As expected, recall and precision decreased with increasing phylogenetic distance to the reference database, but generally, having a representative of the genus in the reference database was sufficient to achieve a relatively high accuracy. We also found variability in the accuracy of identifying individual KOs, with KOs that are more variable in length or in copy number having lower recall. Our paper includes abundance of data on these results, a detailed characterization of the mapping accuracy across different clades, and a description of the impact of additional properties (e.g., read length, sequencing error, etc.).
Importantly, while the obtained functional annotations are in general representative of the true content of the sample, the exact protocol used to analyze the BLAST alignments and to assign functional annotation to each read could still dramatically affect the obtained profile. For example, in analyzing stool samples from the Human Microbiome Project, we found that each protocol left a consistent “fingerprint” on the resulting profile and that the variation introduced by the different protocols was on the same order of magnitude as biological variation across samples. Differences in annotation protocols are thus analogous to batch effects from variation in experimental procedures and should be carefully taken into consideration when designing the bioinformatic pipeline for a study.

Generally, however, we found that assigning each read with the annotation of the top E-value hit (the ‘top gene’ protocol) had the highest precision for identifying the function from a sequencing read, and only slightly lower recall than methods enriching for known annotations (such as the commonly used ‘top 20 KOs’ protocol). Given our lab interests, this finding led us to adopt the ‘top gene’ protocol for functionally annotating metagenomic samples. Specifically, our work often requires high precision for annotating individual reads for model reconstruction (e.g., utilizing the presence and absence of individual genes) and the most accurate functional abundance profile for statistical method development. If your lab has similar interests, we would recommend this approach for your annotation pipelines. If however, you have different or more specific needs, we encourage you to make use of the datasets we have published along with our paper to help you design your own solution. We would also be very happy to discuss such issues further with labs that are considering various approaches for functional annotation, to assess some of the factors that can impact downstream analyses, or to assist in such functional annotation efforts.
Thanks for this … as we get more and more metagenomic data, including from the built environment, this type of analysis will be critical.