The idea for GenomePeek began two years ago when I was working with Karl Klose, Liz Dinsdale, and Rob Edwards to assemble a P. salmonis genome that was being particularly difficult, even though we had 9 gigabases of sequencing. To check whether it was a single isolated genome I pulled out all the 16S reads that hit to 16S and then assembled them. All of the assembled contigs hit to P. salmonis, it was only until later that I found that genome had been shredded by an overactive transposon. We still haven’t solved that problem ….
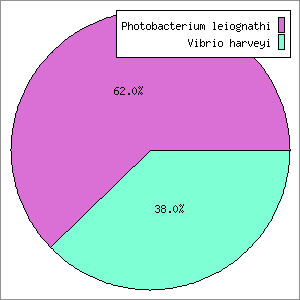
GenomePeek lay dormant for a year, until a student from SDSU’s bacterial sequencing class came looking for help. In the class the students were isolating a bacterium, sequencing, assembling, and then analyzing it. The student wanted to create a recA phylogeny and was having trouble with taxonomic assignment. Their question was: “which RecA gene do I use?”. This set off a red flag immediately, since RecA is essentially a single copy housekeeping gene. On inspection, their assembled genome did indeed have two full copies of RecA; one that hit to a Vibrio and one that hit to a Photobacterium. At the time the two 16S rRNA genes hit to Vibrio, although they were clearly different from each other and hit to different species (since then a representative Photobacterium 16S sequence has been added to the NCBI 16S database that is now the top hit). It turned out that this bacterium also had two sets of other single copy housekeeping genes (I checked rpoB, groEL, nifD, gyrB, and fusA). One of each gene hit to a Vibrio species while the other was most similar to a Photobacterium species. Suffice to say the student was very disappointed after spending a few weeks analyzing and writing up a paper. The idea occurred for a tool where one could submit sequencing data and then quickly get back a set of useful housekeeping genes for phylogenetic analysis. I thought this tool would save everyone’s time wasted on assembly, annotation, and analysis. By quickly checking sequences, we could easily detect whether the original sequencing data was contaminated. I wrote the initial version of GenomePeek, and tested it with various data sets using this student’s data. I quickly found that some genes work better than others, and so currently the four genes analyzed are: 16S, recA, rpoB, and groEL.
One of the things that I wanted to create was a simple user interface so that biologists could easily access the tool. The interface you see was adapted from the BlueImp package. Rob took that and modified it further, which resulted in a massive server breach, however I will leave him to tell that sordid story.
The GenomePeek pipeline has been online a short time but has already been used to find contamination in more student sequencing data, as well as recent sampling data from the Line Islands that we published in PeerJ. Other people have also used GenomePeek to test their sequencing data, and we have also run some samples from NCBI. It is surprising how many “pure genomes” come from more than one organism, especially as we sequence more and more environmental isolates.
Rob was curious whether GenomePeek could be used to analyze metagenomic data and so I analyzed a few sets of artificial metagenomes. GenomePeek is fast, and it appears to have error rates similar to other available metagenomic tools. Of course, each tool is different and provides a different analysis of the metagenome, but the benefit of GenomePeek (aside from its speed) is that since it performs both BLASTN and BLASTX, it has the specificity of a nucleotide homology search and the sensitivity of a protein search. We now use GenomePeek to quickly look at the sequences in our metagenomes.
GenomePeek has also been used in several other unintended applications. The first “disaster averted” was a student having trouble with metagenomic data they obtained. To get an overview of the data I immediately submitted it to GenomePeek, and within a minute we realized the data was 16S amplicon sequencing instead of whole-genome-shotgun sequencing. Of course they were having trouble analyzing that data! When another student was having trouble with metagenomic data we submitted the sequences to GenomePeek, however this time we found that the data was not prokaryotic metagenomes but in fact viral metagenomes. I think this means we need a better way to organize our metagenomes.
GenomePeek is online at http://edwards.sdsu.edu/GenomePeek, and still uses 16S, recA, rpoB, and groEL genes for its analysis. Hopefully when you run it you will see a single organism, but if you are working with environmental isolates don’t be surprised if they are mixed communities.