There is an interesting paper out a few days ago in PeerJ: MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. By , , ,
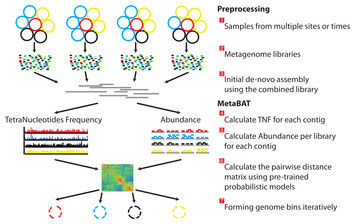
The legend is below:
There are three preprocessing steps before MetaBAT is applied:
- A typical metagenome experiment may contain many spatial or time-series samples, each consisting of many different genomes (different color circles).
- Each sample is sequenced by next-generation sequencing technology to form a sequencing library with many short reads.
- The libraries may be combined before de novo assembly. After assembly, the reads from each sample must be aligned in separate BAM files. MetaBAT then automatically performs the remaining steps:
- For each contig pair, a tetranucleotide frequency distance probability (TDP) is calculated from a distribution modelled from 1,414 reference genomes.
- For each contig pair, an abundance distance probability (ADP) across all the samples is calculated.
- The TDP and ADP of each contig pair are then combined, and the resulting distance for all pairs form a distance matrix.
- Each bin will be formed iteratively and exhaustively from the distance matrix.
So – basically what MetaBAT does is to carry out post-assembly analysis of metagenomic data sets and then bins the contigs from the assemblies using a variety of pieces of information about the contigs. Not 100% sure how useful this is / will be but seems worth trying out for those trying to assemble / bin metagenomic data.