Microbial ecology has benefited enormously from the development of high throughput sequencing technologies, driving the discovery of massive diversity in environments from the ocean to the human body. Where sequencing of less than one hundred 16S rRNA genes from several samples used to be common place with cloning and Sanger sequencing, we can now generate tens and even hundreds of thousands of sequences per sample, from several hundred samples at a similar cost. While some have questioned the need for tens of thousands of sequences for some samples, high throughput sequencing has certainly allowed for much more robust experimental designs for the study of microbial communities, with the ability to simultaneously compare many replicates and conditions.
The obvious trade off of these high throughout platforms has been read length; while Sanger sequencing allows for paired end reads which together cover the entire 16S rRNA gene, current technologies allow anywhere from 250 (Illumina MiSeq) to 500 base pairs (Roche 454) with sufficient quality (i.e. < 0.02% error). Some platforms such as PacBio offer longer read lengths which do allow for full length 16S sequencing, however the error rates are still too high for accurate taxonomic classification and clustering, even after quality filtering.
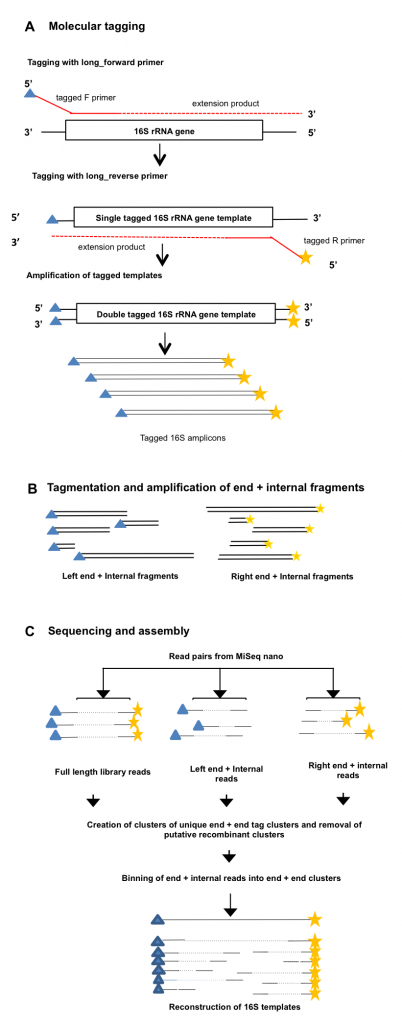
We developed a method to sequence full length 16S rRNA gene amplicons on the Illumina platform. This method is based on adding unique molecular tags to each end of the 16S templates, such that all the sequence information can be reconstructed after fragmentation and paired end sequencing. See Figure 1 for an overview of the protocol.

Molecular tagging of 16S amplicons has been described previously for the purposes of creating consensus sequences from short reads for improved sequence quality. We employed a similar method to tag full length amplicons, by incorporating a string of 10 random nucleotides into both the forward and reverse primers. We used a single extension reaction for each of the forward and reverse primers to tag the 16S rRNA gene templates in our samples, then used additional primers specific to the outer regions of the original primers (Illumina adaptors) to amplify only the tagged templates. We then fragmented the full length tagged amplicons using nextera tagmentation, but altered the PCR step such that we amplified only those fragments which contained one tagged 16S end, and one nextera adaptor end. Paired end sequencing was then carried out on both the full length and fragmented amplicon pools, and the unique molecular tags used to assign short (250bp) reads to sequence bins, followed by assembly of the full length 16S sequence.
We applied this method to DNA from skin swabs, and were able to assemble 2304 sequences, 70% of which were greater than 1300bp, from a single MiSeq Nano run. Assuming linear scaling, we estimate that it is possible to generate up to 80,000 full length 16S sequences on a 600 cycle MiSeq v3 kit, while a HiSeq 2500 might generate more than 480,000 full length 16S sequences in a single “rapid run” lane. We compared this method to 250bp V4 amplicon sequencing from the same samples, and found that our method produced a very similar taxonomic distribution (for phyla and genera level taxonomic assignments). Additionally, the increased coverage afforded by this method resulted in increased average quality scores – giving us very high quality sequences (average PHRED score 68.6 with a standard deviation of 17.2).
Apart from allowing for reconstruction of high quality, full length sequences, molecular tagging offers other benefits. Putative chimeras can be identified as sequences which contain inappropriate combinations of molecular tags (compared to higher abundance sequences). Molecular tagging can also potentially correct against PCR amplification bias, since sequences are reconstructed from original 16S templates that were tagged only once, and regardless of coverage, we count each individually tagged template as having a count of 1. We estimated rates of chimera removal and PCR bias from our skin microbiome data, and we are currently testing the method on mock communities (taking the lead from this nice pre-print by Pat Schloss) for better benchmarking of the method.
So now we know we can do it, what is the utility of high throughput full length 16S rRNA gene sequencing? Intuitively one would assume that you get improved taxonomic classification with longer sequences, and some studies have certainly shown this; although it can also depend on the reference database, classification method, 16S region used and the origin of the samples. Some have suggested that the real bottleneck in accurate classification of 16S rRNA gene sequences (whether long or short) is the completeness of the reference database or training sequences used , in which case the generation of high quality full length 16S sequences for taxa or environments where this information is lacking would be very useful. And for those who are less interested in names, and more interested in phylogeny, longer sequences provide higher resolution for phylogenetic trees.
We’re pretty excited about this method, and we’d love to hear feedback from this community about whether you consider high throughput full length 16S sequencing to be advantageous and why. Do you think this is a useful method for your particular research applications?
Clever idea. I have one question about a potential artifact that I documented occurring in PCR of mixed populations of mRNAs that have SNP differences. The artifact might occur in your protocol at the step where the you generate the second “full-length” strand. First question is do you carry out any PCR after that step? Don’t recall now how the nextera process works but any PCR after the second strand synthesis could result in the formation of heteroduplexes between closely related products after denaturation which could then be resolved in a later step to either side of the heteroduplex in effect scrambling the SNPs among the rDNA products. But maybe I missed something here. I intend to try this because we have an immediate use for the potentially higher resolution afforded. Out of curiosity how long can you go with the first steps? is 1.3kb pushing it or could one go say VERY long – perhaps 20Kb or more?
Those artifacts (chimeras) are definitely a problem for amplicon sequencing generally. The use of the dual molecular tags with this method allows us to estimate which sequences are chimeric by looking for tags at one or the other end of the amplicon which are already present in a much more abundant amplicon, so we think we are able to remove many of these chimeras. In terms of the length, you are limited by the length of fragments you can cluster on the MiSeq, so that you can get the end+end reads from the full length amplicons to link the molecular tags together. We’ve had success with 1.5kb, but I’m not sure how much higher you can go before you lose a lot of the sequencing potential of the flowcell, as the larger fragments don’t cluster anywhere near as efficiently as the smaller ones. I guess an alternative approach would be to try mate-pair sequencing for the full length amplicons (circularisation to bring the two ends together) but I heard this process is very inefficient, and you would need a different adaptor design.