
Blog post prepared jointly by Andrew Doxey (@acdoxey) and Josh Neufeld (@joshdneufeld)
The “aquariome”
Back in 2013, as part of a project assessing aquarium microbial communities and their role in nutrient cycling, Laura Sauder (graduate student in the Neufeld lab) sequenced a shotgun metagenomic library from a freshwater aquarium biofilter that was installed on this aquarium in Josh Neufeld’s office. Well, there were two metagenomes actually, but only one from a sample that was size selected first to remove larger cells.

Why sequence the aquarium filter metagenome? Because previous studies in the lab identified novel thaumarchaeotes as contributors to nitrogen cycling in aquaria and wastewater. As a collaboration between the Neufeld and Doxey labs, a goal was to assemble thaumarchaeal genomes present in the aquarium biofilter, in advance of Laura simultaneously cultivating those same archaea. Although the assembly was made difficult by low relative abundance of thaumarchaeota, in comparison to bacteria, annotation of the metagenomic libraries was another possible goal.
Comparative metagenomics with only one sample
We knew that the simplest and most straightforward analyses for metagenome characterization was a KEGG pathway analysis. To accomplish this, all genes in the metagenome were essentially compared against the KEGG metabolic pathway database with BLAST, in order to predict the presence/absence of different encoded reactions or pathways. These kinds of analyses are commonplace in metagenomics papers.
Unsurprisingly, such a full pathway analysis identified… well, lots of pathways. So, to make things a little more informative, we decided to annotate reads taxonomically (again, using BLAST), then split them into two bins corresponding to archaeal or bacterial pathways. We then did the KEGG pathway analysis separately on these two groups and, because of this comparative tweak to the analysis, we found something intriguing.
By colouring the KEGG metabolic pathway map blue for shared reactions (found in both archaeal and bacterial reads), red for bacteria-specific reactions, and orange for archaea-specific reactions. Most of the map was shared, or bacteria-specific, as we expected. However, there was a small orange-colored archaeal region of the map that corresponded to the vitamin B12 synthesis pathway. In other words, the KEGG analysis suggested a previously unknown role for thaumarchaeota in vitamin B12 synthesis.
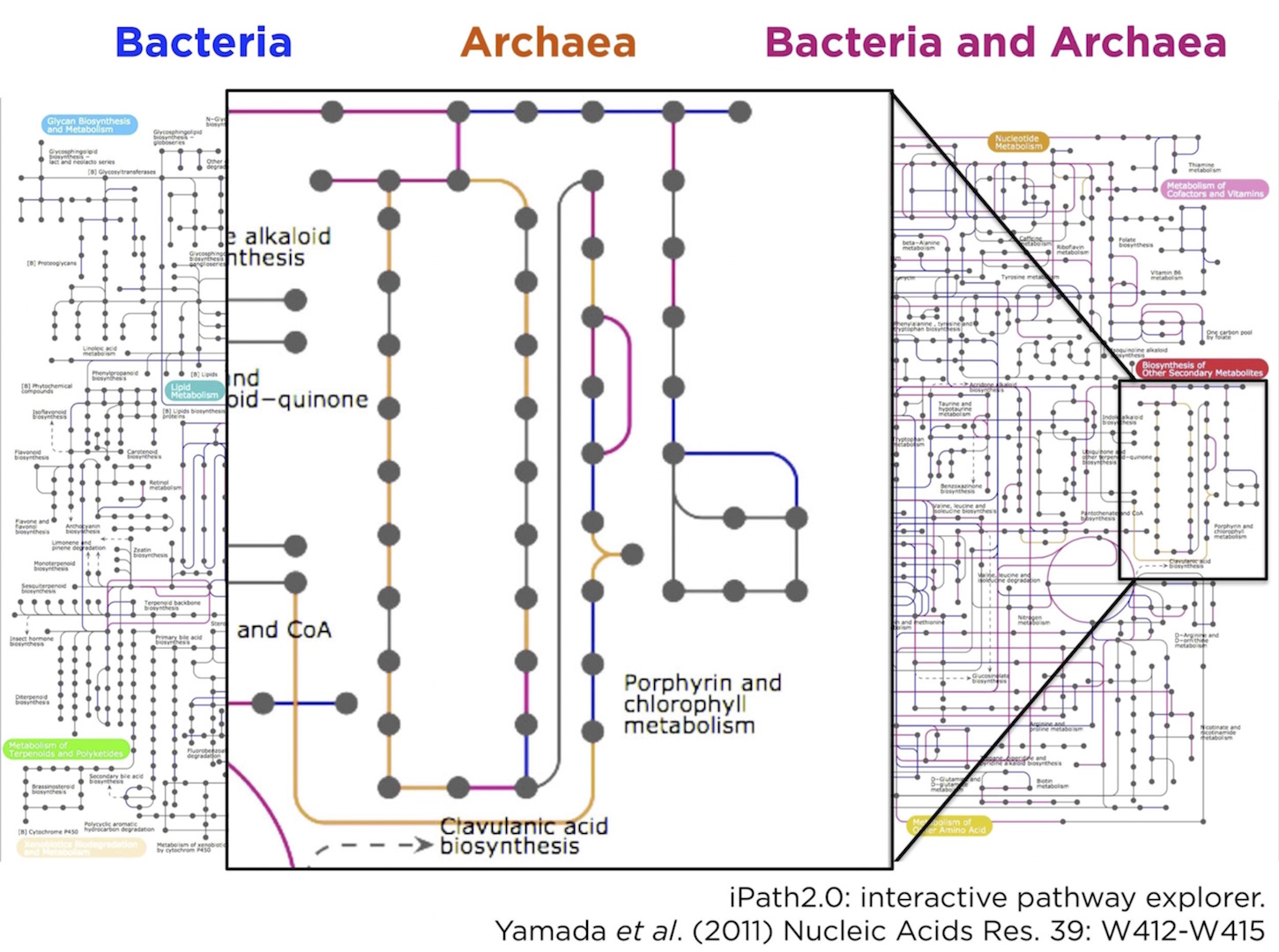
A need for function-specific taxonomic profiling
Realizing that this initial survey of a metagenomic library might have revealed insight into a previously unreported aspect of thaumarchaeal physiology, the next step was to leave the aquarium metagenome behind, for a time, looking instead to see whether this observation was consistent among other datasets from genomes and global sources. We downloaded thaumarchaeal genomes and hundreds of aquatic metagenomes with the goal of reproducing this analysis. However, there was one big problem. It didn’t make sense, nor was it feasible, to do a full KEGG analysis on all 430 metagenomes (actually, 860 metagenomes when dividing them into archaeal and bacterial sequences). Instead, we only wanted to identify the taxonomic contributions to vitamin B12 synthesis genes – who makes vitamin B12 and where?
So, what we decided to do was first “search”, and then “classify”. That is, search for vitamin B12 synthesis genes in all metagenomes and then classify the hits taxonomically. We wrote a pipeline to do this, based largely on BLAST, and found that the thaumarchaeota are indeed dominant contributors of vitamin B12 synthesis in many global aquatic habitats, at least based on genome evidence and relative abundance in metagenomic datasets.
MetAnnotate
Following our vitamin B12 survey, we realized that existing tools for function-specific taxonomic profiling could be improved and automated. Pavel Petrenko, Briallen Lobb, and Daniel Kurtz, all in the Doxey lab at the time, led the development of MetAnnotate (recently published in BMC Biology) to allow users to select any functions of interested that may be defined by GO terms or Genome Properties, for example. This then automatically defines a group of hidden Markov models (HMMs) that represent different protein families for that function/pathway. Individual HMMs can be selected for one-off analyses just as easily from the RDP (FunGene), or other sources. Each of these HMMs are then searched against the user’s metagenomes of interest, and only the hits are taxonomically annotated through closest reference alignment match or phylogenetically. By only focusing on selected HMMs, large comparative analyses can be performed quickly. In fact, in about 10 minutes on an 4-core Linux workstation, we can generate taxonomic profiles of vitamin B12 synthesis genes across a range of environments, as shown below.
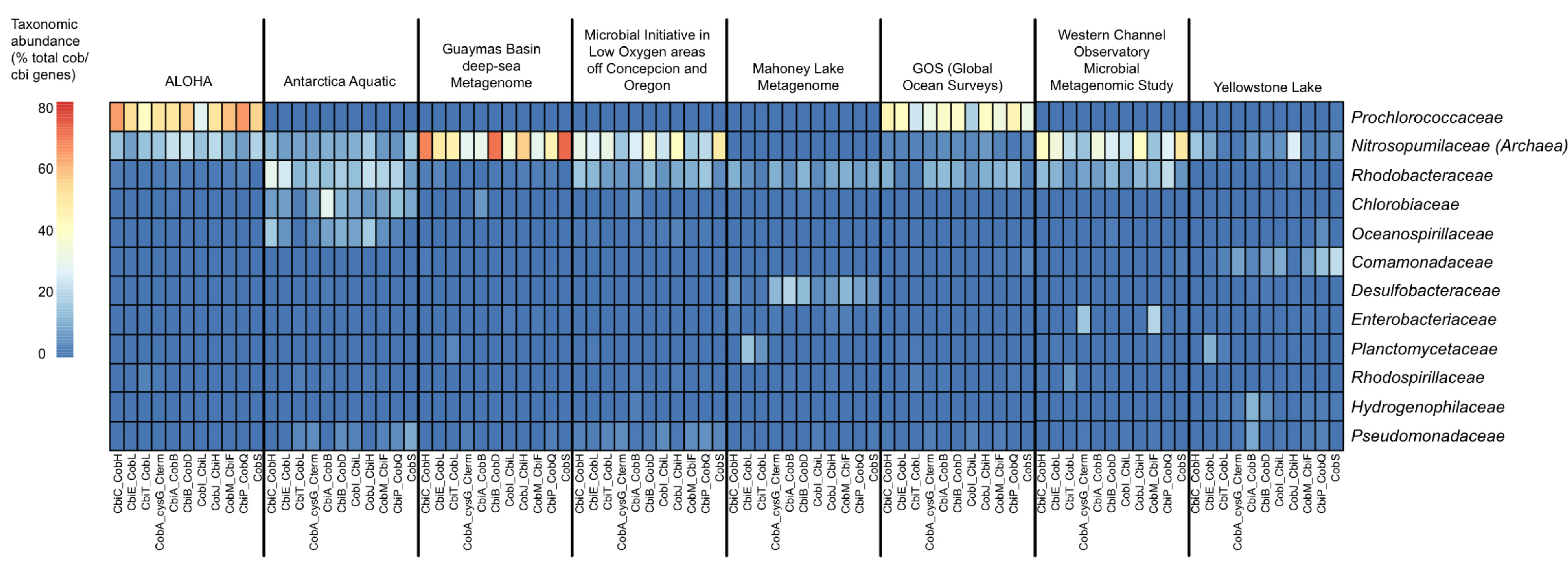
Some of MetAnnotate’s features include:
- Taxonomic profiling of metagenomes using protein species markers
- Taxonomic profiling of metagenomes using custom proteins
- Taxonomic profiling of pathways and GO functions
- Comparative analysis of microbial communities
- Phylogenetic analysis of metagenomes
- Metagenome homology search
MetAnnotate can be installed as a local command-line tool or as a web-server. The project homepage is metannotate.uwaterloo.ca and the source code is available here.
What’s next?
We have plans for MetAnnotate. Exploring multiple metagenomes remains an important goal of microbiome analyses, and we see an expanded role for our tool in making functional sense of sequence data. One future development to look for involves accurate protein subfamily classification, built-in methods for detecting significant differences between datasets, and statistics for highlighting species that contribute disproportionately to functions of interest compared to that expected from their community abundance.
And what about our aquarium filter metagenomes? Stay tuned. We’re working on a report that will focus on vitamin B12, ammonia-oxidation, nitrite oxidation, and other genomic insights into this unique and high-biomass habitat of the built environment.