Why use Bracken instead of Kraken?
by Jennifer Lu and Steven Salzberg
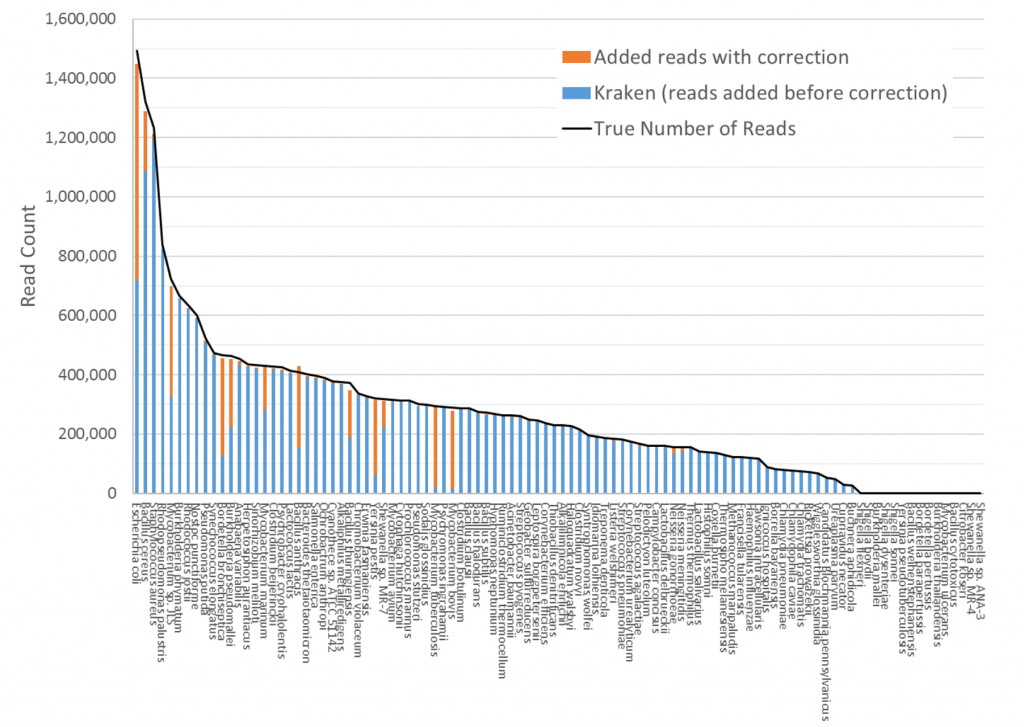
Kraken is a very fast, accurate program for classifying metagenomic sequencing data. It takes a set of reads, contigs, or other DNA sequences and for assigns a taxonomic label (species, genus, etc.) to each one. Last year we discovered that some people were using Kraken directly for abundance estimation – for estimating the relative proportions of species in a sample – and were publishing papers based on the assumption that Kraken’s output can be used this way. However, this is incorrect. If you give Kraken a set of metagenomic reads to classify, it will assign to each read the most specific label it can. Many times, though, these labels are not at the species level. For instance, if a 150bp read is 100% identical to two different species, Kraken will assign it to their lowest common ancestor (LCA), which could be at the genus level or higher. For a sample containing two or more highly similar species, this means that the number of species-specific reads may be far less than expected. (We should note that Kraken often assigns reads at the strain level as well.)
To address this issue, we developed Bracken: Bayesian Re-estimation of Abundance after Classification with KrakEN. Bracken uses a Bayesian algorithm and the Kraken classification results to estimate species-level or genus-level abundances for a metagenomic sample. This new tool is described in detail in our PeerJ paper, https://peerj.com/articles/cs-104/ or on the Bracken software website, https://ccb.jhu.edu/software/bracken/.
Bracken turns the Kraken output into estimates of species-level abundance. This is especially useful when highly similar species occur within the same sample. For example, the genomes of Mycobacterium bovis and Mycobacterium tuberculosis are 99.95% identical. Kraken therefore would classify the vast majority of reads from either of these species as the genus Mycobacterium (because they are indistinguishable). However, some reads within a particular sample usually come from the unique (or species-specific) portion of the genome. Bracken uses this information, plus information about the similarity between the sister species, to “push down” reads from the genus level to the species level. If you are interested in knowing how much of each Mycobacterium species exists within the sample, you can then use Kraken followed by Bracken to provide an estimate of the relative abundance of each species.