A new preprint has come out in PeerJ that presents a simple yet elegant solution to one of the bigger problems in DNA based microbial diversity studies. The authors (Wenke Smets, Jonathan W Leff, Mark A Bradford, Rebecca L McCulley, Sarah Lebeer, Noah Fierer) show how one can use the addition of an internal standard prior to DNA extractions to allow for the estimation of actual (i.e., not relative) abundances of organisms in samples. Really interesting and simple and apparently effective. Abstract and paper details are below:

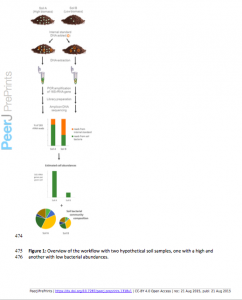
Many recent studies rely on 16S rRNA-based sequencing approaches to analyze bacterial or archaeal communities found in soil and other environmental samples. While this approach is valuable for determining the relative abundances of different microbial taxa found in a given sample, it does not provide information on how the total abundances of targeted microbes differ across samples. Here we demonstrate how the simple addition of an internal standard at the DNA extraction step allows for the quantitative comparison of taxon abundances across samples. The reliability of this method was assessed in two ways. First, we spiked a dilution series of two different soils with internal standards to ascertain whether we could accurately quantify differences in cell abundances. We tested two different internal standards, adding DNA from Aliivibrio fischeri or Thermus thermophilus, bacterial taxa unlikely to be found in soil. Both standards allowed us to accurately quantify microbial abundances in soil as there was a strong positive correlation between total 16S rRNA gene estimations based on the recovery of sequences from these internal standards and the different starting amounts of soil extracted. We then tested whether we could use this approach to quantify differences in microbial abundances across a wide range of soil types. Microbial biomass of these samples was determined with standard methods: phospholipid fatty acid (PLFA) analysis and substrate induced respiration (SIR) analysis. The taxon abundances estimated with the internal standard sequencing approach were significantly correlated with the independent biomass measurements, and were in fact better correlated to SIR and PLFA estimates than either of these two biomass measurements were correlated with one another. Together, these results demonstrate that adding a DNA internal standard to soil or other environmental samples prior to DNA extraction is an effective method for comparing absolute bacterial cell abundances across samples. Given the ease of adding DNA internal standards to soil samples prior to high-throughput marker gene sequencing, absolute abundances and community composition can now be determined simultaneously and routinely.
This is actually quite beautiful…I think more and more titrated molecules will help all these studies, as with the ERCCs in RNA: http://www.nist.gov/mml/bbd/ercc.cfm.