(…) A trial with prospective monitoring of the intensive care unit (ICU) patient microbiome using culture-independent techniques to confirm and characterize this dysbiosis is thus urgently needed. Characterizing ICU patient microbiome changes may provide first steps toward the development of diagnostic and therapeutic interventions using microbiome signatures. (…) Our results demonstrate that critical illness leads to significant and rapid dysbiosis. Many taxons significantly depleted from ICU patients versus AGP healthy controls are key “health-promoting” organisms, and overgrowth of known pathogens was frequent. Source compositions of ICU patient samples are largely uncharacteristic of the expected community type. Between time points and within a patient, the source composition changed dramatically. Our initial results show great promise for microbiome signatures as diagnostic markers and guides to therapeutic interventions in the ICU to repopulate the normal, “health-promoting” microbiome and thereby improve patient outcomes.
System layout of the Vermont, Vietnam Memorial and Visitor Center ecological WWTP.
(…) It is clear conventional wastewater treatment technologies are not meeting the challenge of the mounting pressures on global freshwater supplies. Cost-effective ecological wastewater treatment technologies have been developed in response. To determine whether the removal of micropollutants in ecological wastewater treatment plants (WWTPs) is promoted by the plant-microbe interactions, as has been reported for other recalcitrant xenobiotics, biofilm microbial communities growing on the surfaces of plant roots were profiled by whole metagenome sequencing and compared to the microbial communities residing in the wastewater. (…) Collectively, the microbial communities residing in the wastewater were taxonomically and metabolically more diverse than the immersed plant root biofilm. However, greater heterogeneity and higher relative abundances of xenobiotic metabolism genes was observed for the root biofilm.
Disinfection processes aim at reducing the number of viable cells through the generation of damages in different cellular structures and molecules. Since disinfection involves unspecific mechanisms, some microbial populations may be selected due to resilience to treatment and/or to high post-treatment fitness. In this study, the bacterial community composition of secondarily treated urban wastewater and of surface water collected in the intake area of a drinking water treatment plant was compared before and 3-days after disinfection with ultraviolet radiation, ozonation or photocatalytic ozonation. The aim was to assess the dynamics of the bacterial communities during regrowth after disinfection. (…) Genera such as Pseudomonas, Acinetobacter or Rheinheimera presented a selective advantage after water disinfection. These variations were not observed in the non-disinfected controls. Given the ubiquity and genome plasticity of these bacteria, the results obtained suggest that disinfection processes may have implications on the microbiological quality of the disinfected water.
Diagram of samples collected for genome sequencing of field isolates of MDV.
The intensification of the poultry industry over the last sixty years facilitated the evolution of increased virulence and vaccine breaks in Marek’s disease virus (MDV-1). (…) we developed methods for obtaining high quality genome sequences directly from field samples without the need for sequence-based enrichment strategies prior to sequencing. We applied this to the first characterization of MDV-1 genomes from the field, without prior culture. (…) We found that viral genomes from adjacent field sites had high levels of overall DNA identity, and despite strong evidence of purifying selection, had coding variations in proteins associated with virulence and manipulation of host immunity. Our methods empower ecological field surveillance, make it possible to determine the basis of viral virulence and vaccine breaks, and can be used to obtain full genomes from clinical samples of other large DNA viruses, known and unknown.
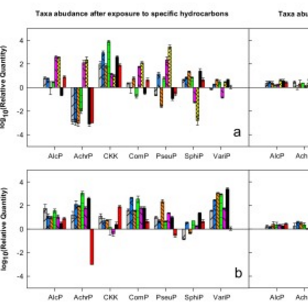
Relative quantity values of hydrocarbon-perturbed cultures
It is not known whether diesel-degrading bacterial communities are structurally and functionally robust when exposed to different hydrocarbon types. Here, we exposed a diesel-degrading consortium to model either alkanes, cycloalkanes or aromatic hydrocarbons as carbon sources to study its structural resistance. The structural resistance was low, with changes in relative abundances of up to four orders of magnitude, depending on hydrocarbon type and bacterial taxon. (…) However, despite this low resistance, structural and functional resilience were high, as verified by re-exposing the hydrocarbon-perturbed consortium to diesel fuel. (…) Thus, the consortium is expected to cope with short-term exposures to narrow carbon feeds, while maintaining its structural and functional integrity, which remains an advantage over biodegradation approaches using single species cultures.
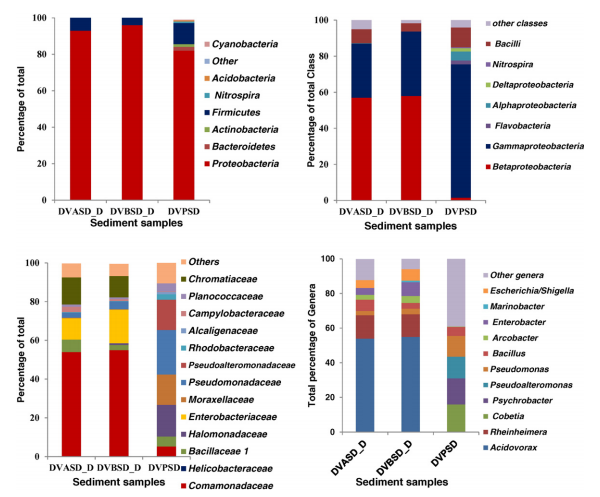
Bacterial community structure from polluted and pristine sediments.
The Alang—Sosiya shipbreaking yard (ASSBY) is considered the largest of its kind in the world, and a major source of anthropogenic pollutants. The aim of this study was to investigate the impact of shipbreaking activities on the bacterial community structure with a combination of culture-dependent and culture-independent approaches. In the culture-dependent approach, 200 bacterial cultures were isolated and analyzed by molecular fingerprinting and 16S ribosomal RNA (r-RNA) gene sequencing, as well as being studied for degradation of polycyclic aromatic hydrocarbons (PAHs). In the culture-independent approach, operational taxonomic units (OTUs) were related to eight major phyla, of which Betaproteobacteria (especially Acidovorax) was predominantly found in the polluted sediments of ASSBY and Gammaproteobacteria in the pristine sediment sample. The statistical approaches showed a significant difference in the bacterial community structure between the pristine and polluted sediments. (…)