(This blog post was prepared by students enrolled in the Koala Poop Microbiome Class in the Fall of 2016 at UC Davis)
This week in lab we finally identified our microbes! The past six weeks have been a culmination of lab work, both successful yet sometimes frustrating, to bring us to this point of finally identifying our isolated microbes through a process known as Sanger Sequencing.
Last week we had performed PCR cleanup on our DNA samples with the intent of removing any “non-DNA” substances from the sample. By removing non-DNA substances, our DNA samples presumably had less “junky” DNA sequence before they were run through Sanger Sequencing. We should be more likely to find a match for our unknown DNA samples in genetic databases without “junk” DNA sequences.
So, what is Sanger sequencing and why do we use it? Sanger sequencing provides us with the building blocks of DNA, sequences of nucleotides, which can then help us identify the organism our DNA sample is taken from. Huge databases of genetic sequences have been created, which are used as a standard by which novel species’ DNA can be compared to.
This video animation further illustrates the process of Sanger sequencing.
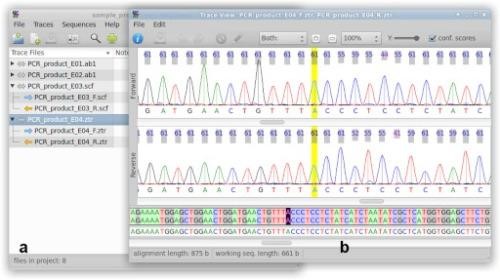
In this lab, we used SeqTrace program to create the consensus (final) sequence by matching forward and reverse sequencing reads. To match the two reads, we cut off the end parts that had not been identify well. Then matched the two sequences with their base-pair to make sure the base-pair that identified was correct in both forward and reverse reads. In other words, ideally, forward and reverse reads should be the same. (Img. 1)
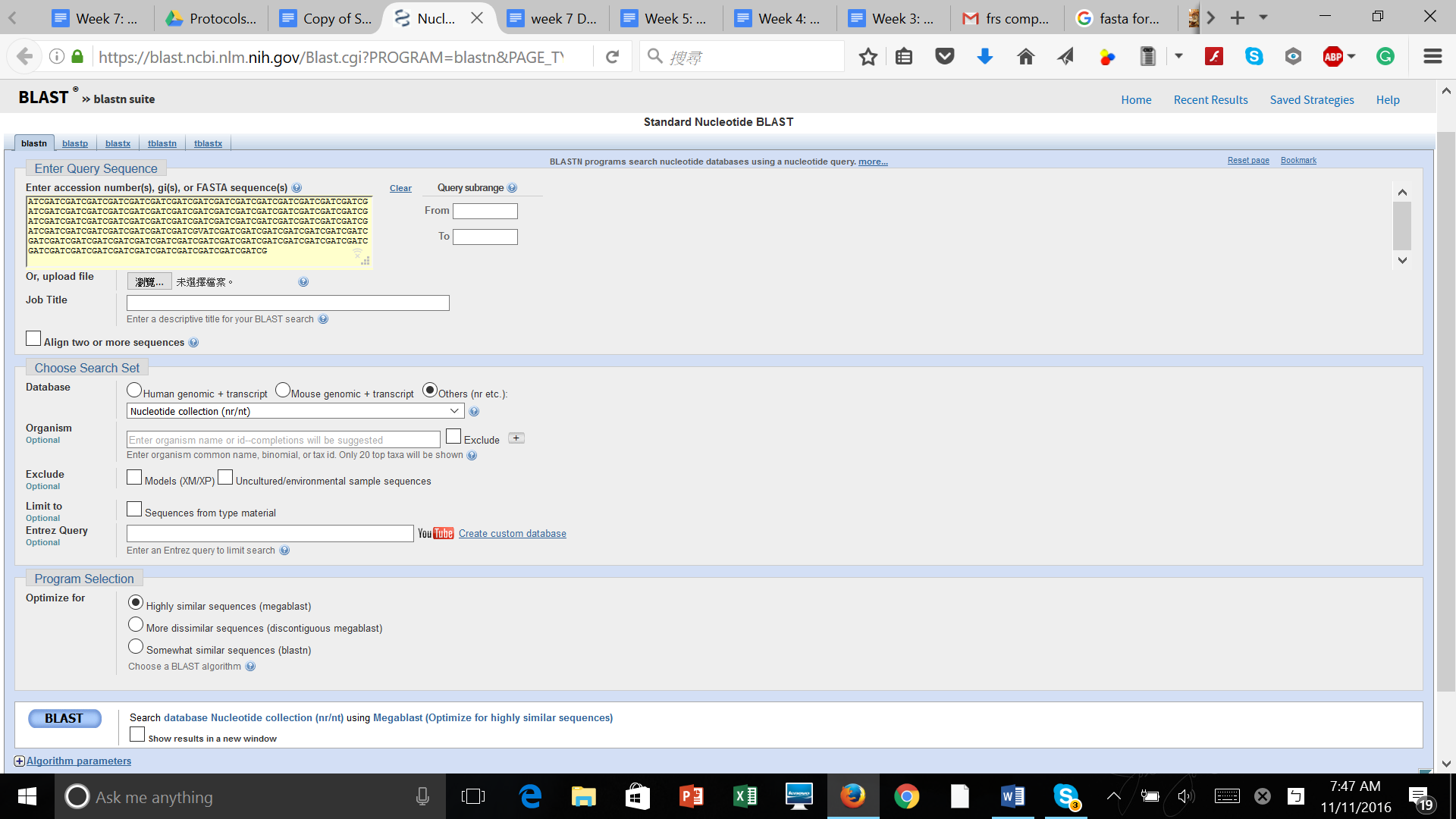
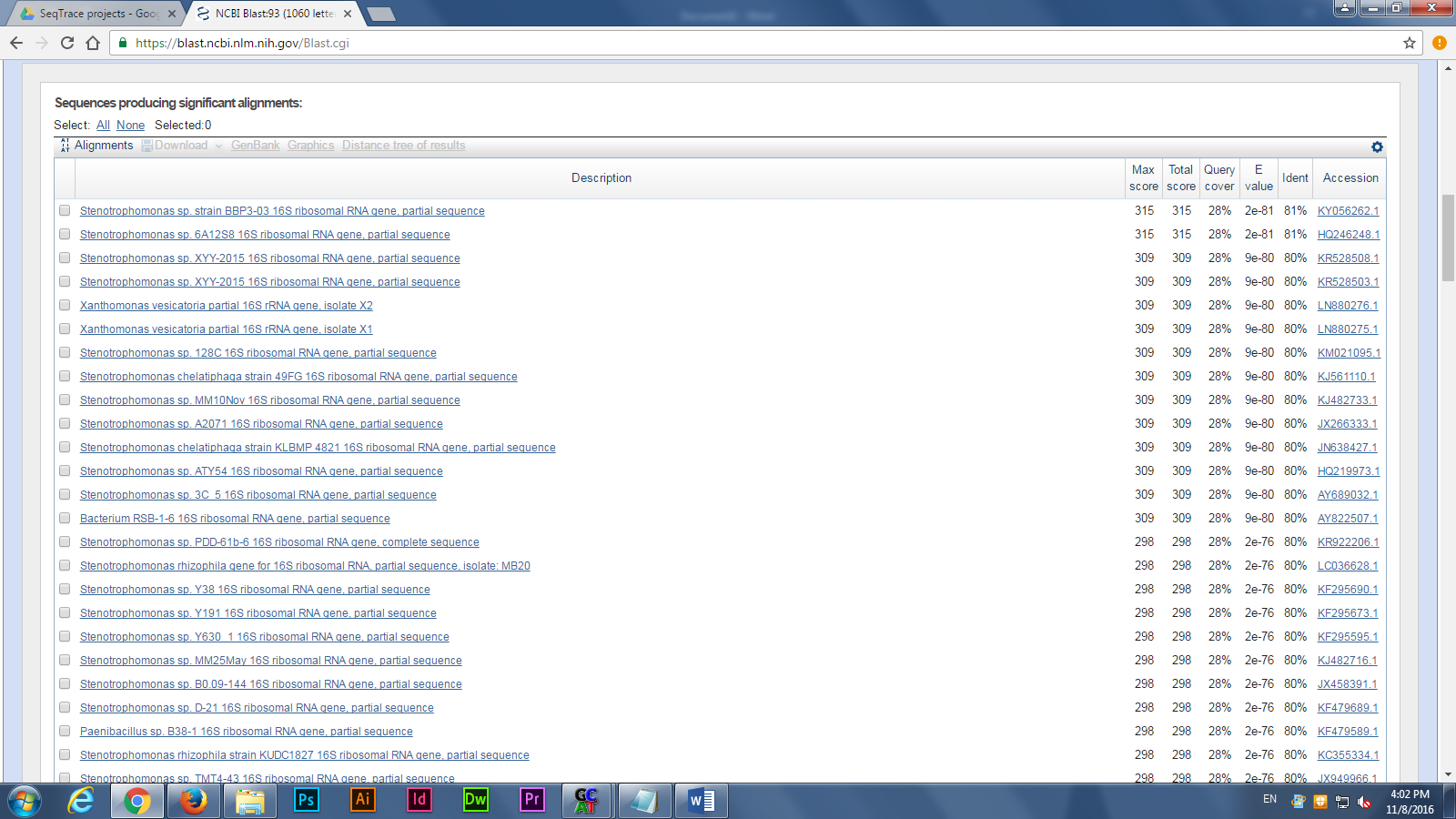
After produced the consensus sequence, we inputted the sequences of nucleotides in FAST format to the BLAST program, database of nucleotides sequences, to identify the possible species. (Img. 2 &3)
Next, we uploaded our nucleotides sequences to RDP program, 16S rRNA sequences analyzing tool, for next week’s lab, building a gene tree to find the closest relation of the possible species. The majority of microbes were identified as various strains of Escherichia coli, which makes sense since E. coli make up a good portion of the microbial community in mammal guts.
The following is a list of some of the many other types of microbes we had in our koala faeces:
Shigella: From the Enterobacteriaceae family and possibly responsible for dysentery
Escherichia fergusonii: Known to infect open wounds in humans, possibly causing urinary tract infections and are highly resistant to ampicillin
Escherichia vulneris: Non-pathogenic bacteria that colonizes open wounds
Lonepinella koalarum: A novel tannin-degrading bacteria
Stenotrophomonas moltophilia: Bacteria that colonizes humid environments and is pathogenic in immunocompromised humans and mammals
Many of our samples did not work, however, which was frustrating since we have put in so much time to prepare our samples for sequencing. Nevertheless, we found it interesting to put a name to some of our mystery microbes and finally see some success from the past weeks of lab work.
Question for science community: What cause some sample forward and reverse base-pair sequences cannot perfectly match each other? Why some same spot has two peaks in SeqTrace graph that represent two possible different nucleotides? Is Sanger Sequencing the most effective way for identify microbes? If no, what kinds of method will it be? In this class, we found the rate of success experiment is a bit low. How to decrease the rate of failure in each steps for class design?
Fun Fact: Sanger sequencing was developed by Frederick Sanger and colleagues in 1977 and is still a widely used sequencing method in small-scale sequencing projects.