

(Note: This post was updated on 3/26/18 after re-running some of our analysis based on suggestions in the comments and on Twitter).
We were very excited to get our MinION sequencer from Nanopore 1.5 years ago… so many potential applications; sequencing in real time, use in the classroom, use in the field, not having to wait to get bacterial genomes, etc. But one thing lead to another and it languished in a drawer. Recently we got it back out and tried to sequence a bacterial genome with it. This project is being led by two undergraduates in the lab, Marcus Cohen and Dennett Rodriguez, to whom I basically gave the MinION with the instructions of “figure out how this thing works”.
First observation: This was way harder than we thought. There are many different protocols/suggestions/approaches/ideas/optional steps. After a month, we managed to generate and then basecall some sequence, then assemble it with Canu into a single contig. Cool! Then it gets complicated.
So I thought it was a simple question that I threw out to Twitter, “So we just assembled our first ever @nanopore bacterial genome. Not understanding how we can get only 65% completeness (CheckM) with 100X coverage. We used an entire flowcell.”
This tweet apparently touched a nerve, starting a wide-ranging discussion about the merits of Nanopore versus Illumina versus PacBio and the utility (or not) of finished (or even decent quality) genomes. There were a few hundred tweets generated, by many of the experts in the field in additions to employees of at least one of the companies. The discussion was surprisingly combative, even degenerating into name calling at one point. However, it was extraordinarily informative to a place like our lab where we do some, but not a ton, of this kind of work.
I couldn’t figure out a good way to collect or archive the tweets which is a shame. But here’s my take home messages from the discussion.
-Nanopore data alone is insufficient to get a decent bacterial genome, due to a high rate of error (homopolymer indels).
-These errors cause frameshifts which lead to genes looking like pseudogenes and renders programs like CheckM (which looks for proteins) basically useless
-Pacbio data alone is better than Nanopore… some people think it sufficient for a finished genome and others disagree and think we always need Illumina plus long reads
-Obviously Illumina data alone is insufficient for closing genomes, but for many applications is quite sufficient
-Hybrid Illumina/PacBio or Illumina/Nanopore data is clearly the best approach for getting good genomes, combining accuracy with long reads. The relative cost of these various approaches is highly debatable
-Both PacBio and Nanopore data require extensive polishing and correction to be usable alone… it seems like these workflows are much more established for PacBio to date
-If I had one bacteria that I wanted a really good genome for, I would probably do PacBio (or PacBio/Illumina if I really really cared), if I had a dozen I would probably do Nanopore/Illumina (because of cost), if I had 100 I would just do Illumina and call it a day.
-Other good uses for Nanopore/PacBio data alone would be anything with repeats, plasmids, CRISPER arrays
So back to our bacteria. Following a blog post from Mick Watson we went ahead and performed his test “First predict proteins using a gene finder. Then map those proteins to UniProt (using blastp or diamond). The ratio of the query length to the length of the top hit should be a tight and normal distribution around 1.” And… ours looks terrible.
This is borne out by looking at our data… at least in terms of marker 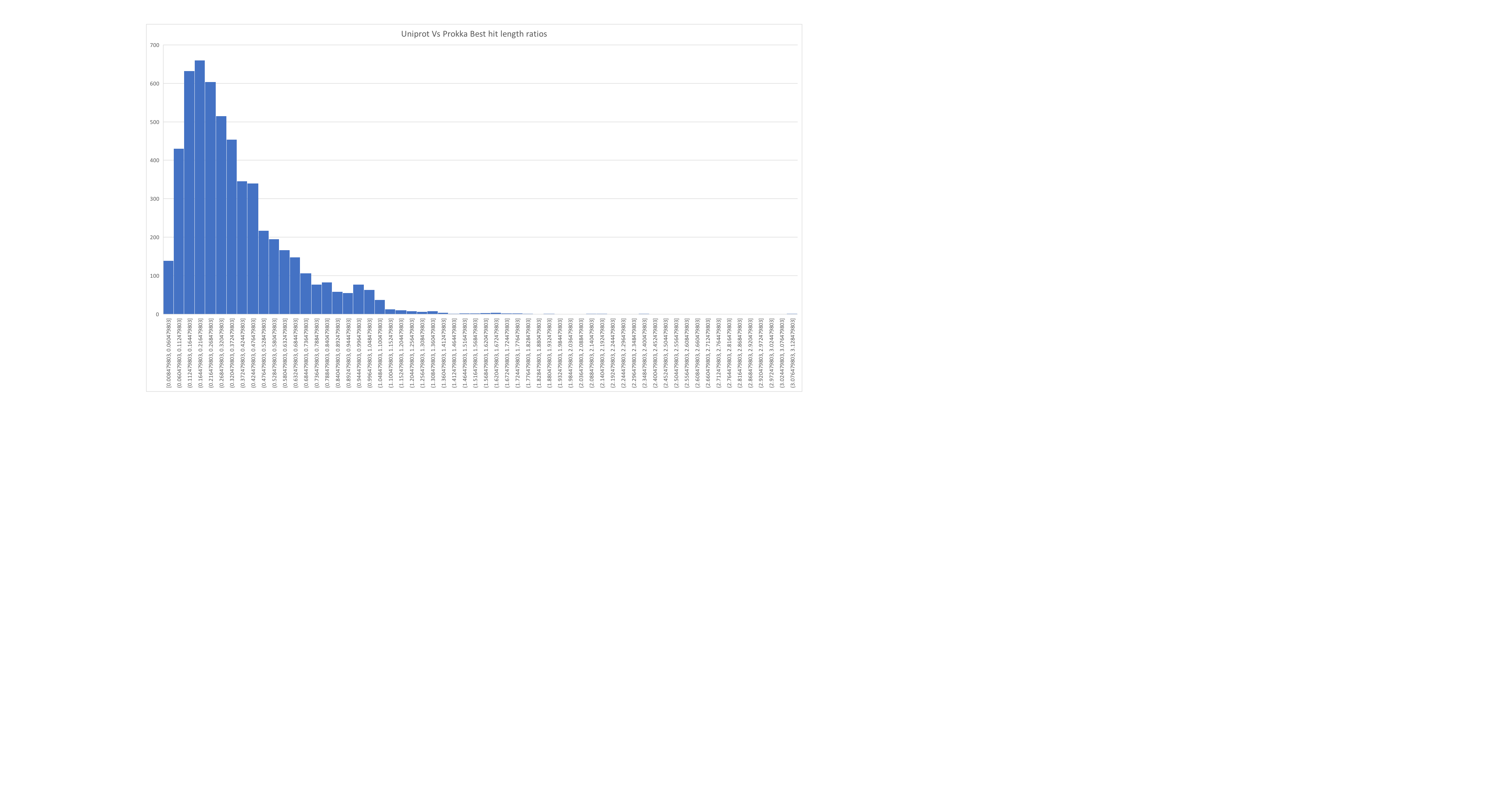 genes and assembly quality. We tried running one round of Nanopolish and that didn’t appear to help.
genes and assembly quality. We tried running one round of Nanopolish and that didn’t appear to help.
So we’re a bit stuck for the moment. Our next steps are to try some more polishing. One of the hardest points is understanding whether our run was just poor or if we’re seeing what’s normal for the technology. We’ve also sent off more DNA for Illumina sequencing so we’ll be trying hybrid approaches next.
UPDATE 3/26/18
After the comments below and some discussion on Twitter, Guillaume re-ran Nanopolish with different parameters and we got much better results… bringing ourselves to 93% completeness instead of 65%. Still not great for many purposes but a huge improvement!
Here’s a Wakelet of the Twitter discussion…
Very cool post David. I am also slowly trying to get up to speed w Nanopore. I think it will be incredibly valuable as a teaching tool (since it brings the whole process to the lab). Input DNA quality seems to be the big sticking point – which is a little bit punny if you’ve used the “ultra-long read DNA prep” method. Lots of cleanup seems to be the order of the day. Order lots of Phenol:Chloroform:Isoamyl ! but I think you are right that hybrid assembly is the way to go – I think the program is called Unicycler (includes both long reads and illumina data). My one try at this has been marginal, but we are trying to get the new baby Illumina machine (iSeq) so we can set up the whole process in house. And yes, the twitter fights are epic – but aren’t they always?
Yeah our next try was supposed to be metagenomics in the field but that seems hard to contemplate because we don’t get much DNA out of most environmental samples.
yeah we tried some metagenome samples. you def need even better cleanup. we had some success in getting decent (but prob not good enough) dna, but gotta keep working at it. the ppl on twitter make it look so easy!
Very nice post David, thank you!
I was wondering if you could elaborate a bit on “We tried running one round of Nanopolish and that didn’t appear to help.”. Did you run the homopolymer correction + the methylation-aware option? From our E. coli experience, it made a substantial difference: # SNPs reduced to about 2.5k and CheckM completeness of ~95%.
We have not yet been able to check for the frameshift/length distribution, though.
Cedric, thanks for the comment! It’s a bit like playing telephone here, but I’ve passed on your comment to our bioinformatician, Guillaume Jospin, who actually runs all this stuff. We’ll let you know.
Thanks for the help Cedric. I ended up starting nanopolish from scratch and got much better results this time. I think I may not have indexed my reads properly last time.
Anyways, the CheckM results came back at 93% completion this time instead of 68(or so before). So that’s great news. I did not use the methylation-aware option. We don’t care about that information but does it help the polishing step??
Here an updated of the distribution of protein length matches
https://ucdavis.box.com/s/b2cvknjm6ifjdwxl5j9zet0hctpmbaqn
Hi David
sometimes multiple rounds of nanopolish used to help, I think now with methylation aware you should reach about ~99.9% identity or you could use chiron to reach a good assembly…. But systematic errors remain (I reckon mostly homopolymers) which can at the moment -at least using public software to my knowledge- not be polished away. This might (or not change) maybe on the london calling nanopore conference.
Have a look at this great resource for bacteria
https://github.com/rrwick/Basecalling-comparison
Best Wishes
Björn
Thanks for the comment. We are revisiting Nanopolish right now and seeing if we’re just doing it wrong.
David. Even though Storify is going away, one can still make new Storifies for a few months. So I made a Storify of the Tweet discussion of this. See https://storify.com/phylogenomics/can-you-sequence. I am in the process of importing this into Wakelet (a good replacement to Storify). But for now you can use the Storify. Don’t say I never did anything for you.
And here is a link to the Wakelet — https://wakelet.com/wake/a24a7449-f57e-45aa-ae93-8e41b6fe8340
Thanks!
note – you could embed either of those (probably better to do the Wekelet)
Done.