This could be of use to many people:
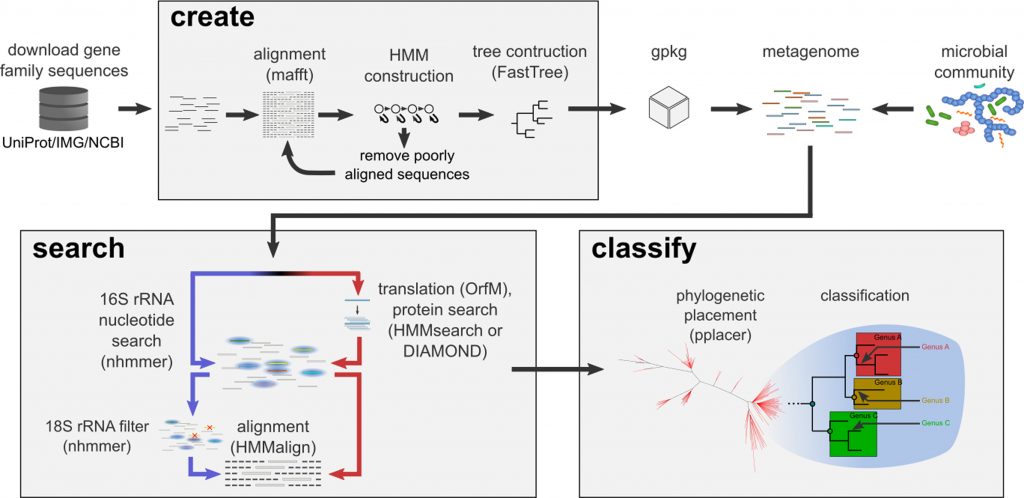
Large-scale metagenomic datasets enable the recovery of hundreds of population genomes from environmental samples. However, these genomes do not typically represent the full diversity of complex microbial communities. Gene-centric approaches can be used to gain a comprehensive view of diversity by examining each read independently, but traditional pairwise comparison approaches typically over-classify taxonomy and scale poorly with increasing metagenome and database sizes. Here we introduce GraftM, a tool that uses gene specific packages to rapidly identify gene families in metagenomic data using hidden Markov models (HMMs) or DIAMOND databases, and classifies these sequences using placement into pre-constructed gene trees. The speed and accuracy of GraftM was benchmarked with in silico and in vitro mock communities using taxonomic markers, and was found to have higher accuracy at the family level with a processing time 2.0–3.7× faster than currently available software. Exploration of a wetland metagenome using 16S rRNA- and methyl-coenzyme M reductase (McrA)-specific gpkgs revealed taxonomic and functional shifts across a depth gradient. Analysis of the NCBI nr database using the McrA gpkg allowed the detection of novel sequences belonging to phylum-level lineages. A growing collection of gpkgs is available online (https://github.com/geronimp/graftM_gpkgs), where curated packages can be uploaded and exchanged.
And kudos for making the software open and the paper open.
