With our paper recently accepted for publication and many months of practical application, we’re excited to spread the word about our new tool for characterizing metagenomes, PALADIN! Short for Protein ALignment And Detection INterface, PALADIN was our response to the inherent problem of limited references in shotgun metagenomic sequencing. While DNA barcoding involving 16S, 18S, and ITS amplicon sequencing creates for faster assembly/alignment (in comparison), the overall technique falls short if the species is poorly represented in the reference database. Compounding this, differing constraints and rates of evolution between these markers and the functional genome, as well as horizontally transferred rRNA genes, can lead to incorrect taxonomic inference, resulting in database hits on divergent species whose corresponding barcode markers have sequence similarity.
Instead, we wanted to stick with characterizing all genes present the microbial community to help eliminate this potential issue (as well as provide a more detailed functional profile). Additionally, we made the decision to perform alignment at the protein level, allowing synonymous coding sequences to increase our potential for database hits on homologous representatives in the database. That decision normally leads to BLAST jobs with unfeasibly huge CPU-hour requirements – completely in contrast to the speed gains in DNA barcoding. Given that reality, our goal became to create an aligner with the efficiency of a nucleotide read mapper, but the functionality of BLASTX – and from this, PALADIN was born.
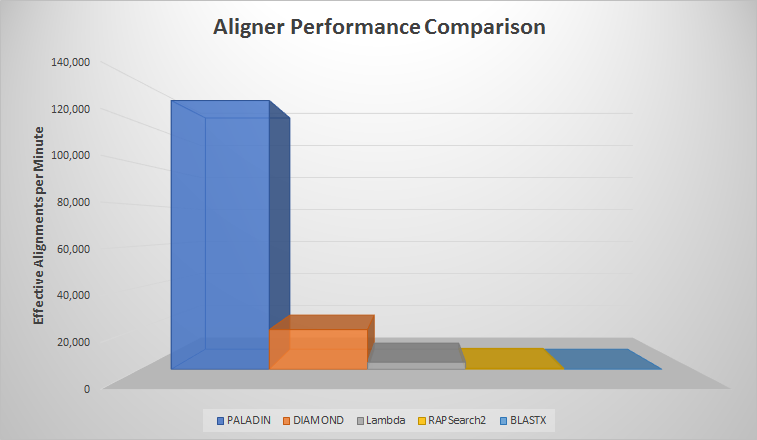
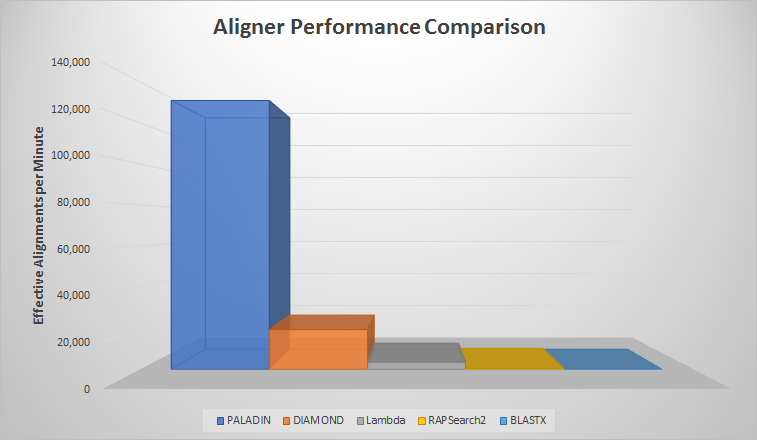
Getting down to brass tacks, PALADIN performs very well in a space that plays host to many BLASTX competitors. Benchmarks show our tool outperforms modern alternatives like DIAMOND, Lambda, and RAPSearch2. The most striking result was against the venerable BLASTX, which took 8000x longer to align than PALADIN – a striking difference!
| Read Count | Aligner | Effective Time (HMS) | Effective Aligns/Min |
|---|---|---|---|
| 1,000,000 | PALADIN | 00:07:10 | 139,535 |
| 1,000,000 | RAPSearch2 | 32:54:59 | 506 |
| 1,000,000 | DIAMOND | 00:51:42 | 19,342 |
| 1,000,000 | Lambda | 04:46:23 | 3,492 |
| 240,000,000 | PALADIN | 31:15:02 | 127,998 |
| 240,000,000 | BLASTX | 250,000:00:00 | 16 |
We achieve this performance by differentiating our tool in the following ways:
- Uses BWA’s BWT/SA indexing and alignment code, a proven and very fast set of algorithms
- Aligns in protein space, inherently improving efficiency over nucleotide alignment by increasing unit atomicity size (three base-4 digits becomes a single base-20 digit)
- ORF detection preparation, reducing the number of non-coding sequences considered during alignment
- Optimized for use with the UniRef-90, thereby leveraging the computational work already performed by UniProt in clustering these sequences
In addition to mapping subject and quality, PALADIN creates a functional characterization using UniProt data, including GO terms, pathway features, and cross references into popular databases such as KEGG, Ensembl, and PATRIC. We’ve had such great results with the tool here at UNH that we’ve also created a framework of accompanying plugins that aids in several downstream tasks, such as an MPI/HPC wrapper for computing cluster execution, mapping present metabolic pathways, taxonomic grouping at any desired rank, GO term abundance, second pass alignment for refining UniRef90 hits to constituent sequences, and custom reports that incorporate any fields available from UniProt.
All in all, we think PALADIN really has the potential to open up new avenues in metagenomic characterization, especially in time and resource sensitive environments. As an actively developed open source tool, we also see a long future of development, which is a critical for sustainability in an area that is unfortunately subject to many abandoned projects. If you’re interested, you can grab PALADIN on Github. If you have any questions or suggestions, please send feedback our way as we continue to improve PALADIN!