Infants born via c-section have a microbiome community composed mostly of skin bacteria [1-3], but the source of these skin bacteria is unknown. People quickly shed bacteria into their environment, leaving their own bacterial signature in a room within hours [4]. Do hospital operating rooms harbor skin bacteria that could colonize c-section delivered infants? A …
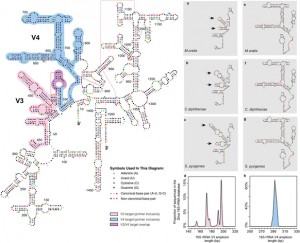
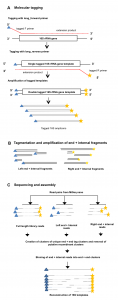
To date, characterization of ancient oral (dental calculus) and gut (coprolite) microbiota has been primarily accomplished through a metataxonomic approach involving targeted amplification of one or more variable regions in the 16S rRNA gene. For anyone interested in rRNA studies of microbial communities, ancient microbiomes, or analysis of samples with small amounts of DNA this …
Gearing up the UNITE database for the built mycobiome The team behind the UNITE database for molecular identification of fungi has been granted support from the Sloan Foundation to strengthen the support for fungi from the built environment. Launched in 2001 as an ITS database for identification of ectomycorrhizal fungi in the Nordic countries, UNITE …
Norm Pace gave a talk at UC Davis yesterday on “Metagenomics and the Tree of Life”. I and a few other people posted live Tweets from the talk which I have compiled together via the Storify system. This “Storify” is embedded below. In addition, Lisa Cohen, who was at the talk posted her notes which …
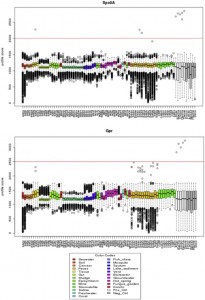
Microbial ecology has benefited enormously from the development of high throughput sequencing technologies, driving the discovery of massive diversity in environments from the ocean to the human body. Where sequencing of less than one hundred 16S rRNA genes from several samples used to be common place with cloning and Sanger sequencing, we can now generate tens …
When I first started trying to do PCR in Colleen Cavanaugh‘s lab in 1989, I was kind of on my own. Colleen was a newly hired profession at Harvard. She was busy getting things set up. And I was the only person in the lab – and I really knew very little. And basically I …
A moderately new paper is out that is an excellent example of how biases in DNA extraction can have major impacts on inferences from culture independent DNA studies. The paper is in what I think is a generally non open access journal (Computational and Structural Biotechnology Journal) for fortunately has been made open access Source: …
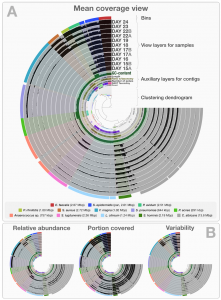
Three weeks ago I stood in front of the 60 attendees of the STAMPS course and asked, “How many of you are currently working with shotgun metagenomes?” Ten to fifteen people raised their hands. In contrast, almost all had their hands in the air when I asked how many were expecting to work with shotgun …
Suppose you owned a warehouse that serves as a distribution hub for grocery stores, and you find that every so often, someone is pooping in your warehouse. Not only is that insulting and obnoxious, but it also has the potential to make a lot of people very sick. You take the shift schedule, and you correlate …
So … what goes around comes around. In 2003 and 2004, I spent a lot of time discussing and arguing with people about what would be the best strategy for making and sequencing Sanger libraries for metagenomic sequencing for the Sargasso Sea metagenome study coordinated by the Venter Institute (I worked at TIGR at the time and …