(This blog post was prepared by students enrolled in the Koala Poop Microbiome Class in the Fall of 2016 at UC Davis) Polymerase Chain Reaction Polymerase Chain Reaction (PCR) amplifies a small amount of DNA into a much larger amount of DNA within a short time. The purpose of this lab was to prepare the …
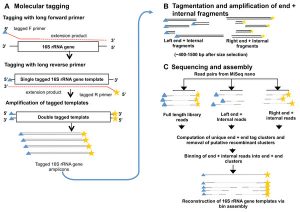
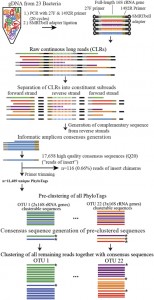
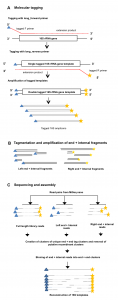
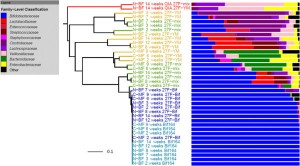
Definitely worth checking this one (from Burke and Darling) out if you do microbial diversity studies … Background The bacterial 16S rRNA gene has historically been used in defining bacterial taxonomy and phylogeny. However, there are currently no high-throughput methods to sequence full-length 16S rRNA genes present in a sample with precision. Results We describe …
This seems like it could be of interest to microbial community researchers: The ISME Journal – High-resolution phylogenetic microbial community profiling Abstract (with bolding by me) Over the past decade, high-throughput short-read 16S rRNA gene amplicon sequencing has eclipsed clone-dependent long-read Sanger sequencing for microbial community profiling. The transition to new technologies has provided more …
Microbial ecology has benefited enormously from the development of high throughput sequencing technologies, driving the discovery of massive diversity in environments from the ocean to the human body. Where sequencing of less than one hundred 16S rRNA genes from several samples used to be common place with cloning and Sanger sequencing, we can now generate tens …
When I first started trying to do PCR in Colleen Cavanaugh‘s lab in 1989, I was kind of on my own. Colleen was a newly hired profession at Harvard. She was busy getting things set up. And I was the only person in the lab – and I really knew very little. And basically I …
Another paper on how sample processing (and in this case PCR primer choice) can influence microbiome studies. And another one that is definitely worth looking at: 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome 2015, 3:26 doi:10.1186/s40168-015-0087-4 by Alan W. Walker, Jennifer C. Martin, …
Well, been having many discussions recently about PCR amplification happening from “negative” controls where no sample DNA was added. Such amplification is alas pretty common – due to contamination occurring in some other material added to the PCR reaction. Obviously it would be best to eliminate all DNA contamination of all reagents and all PCRs. …
One of the issues that was raised in the recent Microbiology of the Built Environment conference in Boulder was sampling, specifically what and how is the material collected for subsequent biological analysis. Industrial hygienists and those tackling questions of exposure have devoted a lot of time to developing methods for how to study the indoor …
Sequencing of PCR-amplified marker regions (e.g. 16S, ITS) for characterization of sample microbial ecology is a widely-used tool in Microbiology of the Built Environmenta (MoBE) investigations. Due to the large amount of data produced by these methods, sequences are typically clustered into operational taxonomic units (OTUs) based on sequence similarity to simplify downstream processing. However, …
Norman R. Pace, from UC Boulder, gave a talk at UC Davis yesterday about microbial diversity. In his talk he discussed some of his recent Sloan Foundation funded work on “microbiology of the built environment” including studies of shower heads, indoor swimming pools, water supplies, and hospitals. Pace is one of the pioneers of DNA …