![Join Indoor Air 2024’s Conference Countdown Challenge]() Join Indoor Air 2024’s Conference Countdown Challenge
Join Indoor Air 2024’s Conference Countdown Challenge
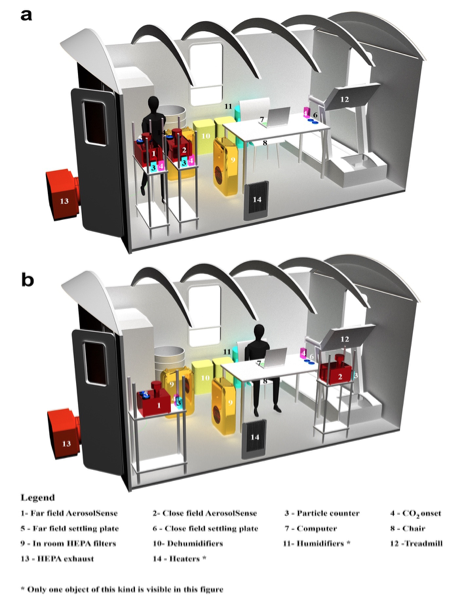
(posting on behalf of Dr. Karen Dannemiller) The Indoor Air 2024 conference from the International Society for Indoor Air Quality and Climate (ISAIQ) will take place in beautiful Honolulu, HI. Have your research highlighted as part of Indoor Air 2024’s Conference Countdown Challenge! Starting July 10th, 2023, submissions of images related to the conference themes ...